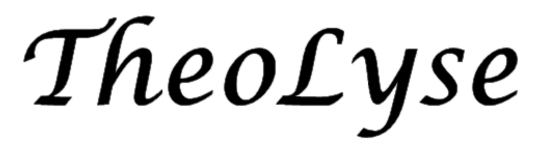(※ここで「従来法」とは系全体を直接計算する既存の量子化学計算手法を指しています)
Elongation法(巨大系量子化学計算法) TS/TB法(相互作用解析法)
背景 従来の非経験的(ab initio)量子化学計算は系サイズの4~7乗オーダーという膨大な計算コストのため、計算機の発達した現在でも高分子材料や生体分子は計算が困難です。計算を可能とするために様々な近似法が提案されていますが、定性的にも間違った結果を与えかねず本質的な解決策にはなりません。比較的小さな分子で無視できる誤差であっても大規模系だと数十~数百kcal/molといった誤差となり、化学精度での議論は到底できません。真の技術革新のためには、望みのサイズの系を近似なく高精度に、しかも常識的な時間・資源で計算できるab initio量子化学計算手法が必要です。
Elongation(ELG)法とは 革新的創薬・材料開発に資する量子化学計算法を目指し、当グループでは超高速と超高精度を兼ね備えた巨大系計算法Elongation法を開発してきました[1,2]。本方法は、高分子の重合反応を模倣し、反応末端へのモノマー付加を繰り返して電子状態を伸長させます(下図左)。反応末端だけを真面(まとも)に、しかも反応末端以外の部分の電子状態もきちんと含めつつ計算します。ここでは軌道を領域に局在化するというオリジナル手法が盛り込まれており、計算精度を落とすことなく大きな系の計算が可能です。
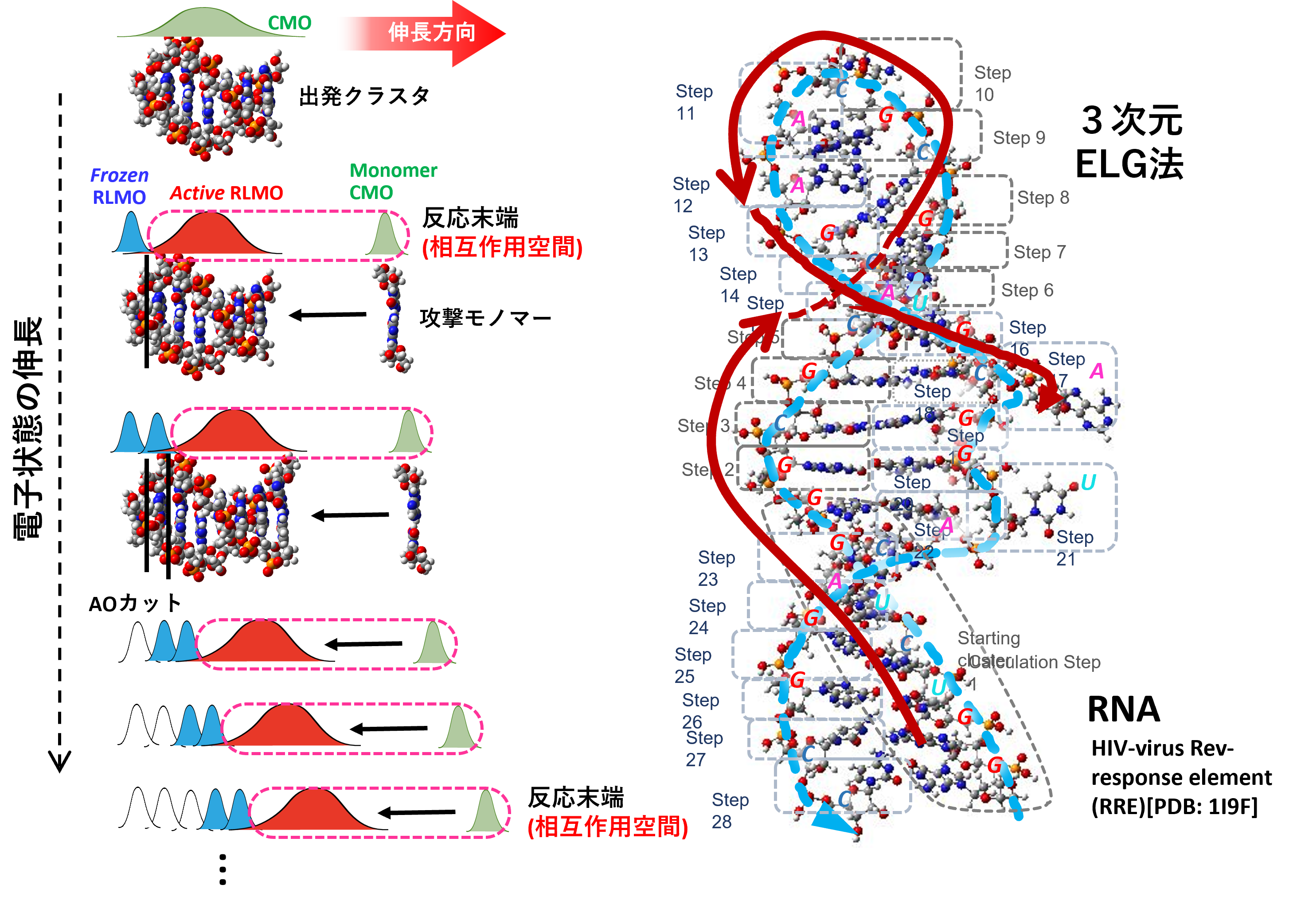
Terminal-to-center(T2C)ELG法[3]は、両末端から中央方向に計算レベルを上げつつ電子状態を伸長します。高分子-リガンド結合系のような大規模系中の局所解析に威力を発揮し、高分子部分を一度だけ計算してデータを保存しておけば、リガンド付加の最終ステップのみ分子を変えながら繰り返すことで、様々なリガンドとの結合エネルギー等をさらに効率的に獲得できます。
機械学習と好相性なELG法 ELG法、T2C-ELG法ともにその逐次計算という特徴からデータベース作成に適しています。本手法ならではの「高分子の電子状態データベース」が機械学習の教師データとしても有効であることをすでに確認しています。
背景 有機化学の分野でThrough Space/Through Bond相互作用という言葉を使った解釈がしばしばなされていますが、その相互作用の大きさを定量的に見積もる方法はなく、結果からの類推の域を超えていないのが現状です。非経験的レベルで軌道相互作用を解析する方法は提案されているものの、その見積もりの正確さ、電子相関効果の取り込みなど定量性の観点から十分とは言えず、いまだ確立した方法はありません。
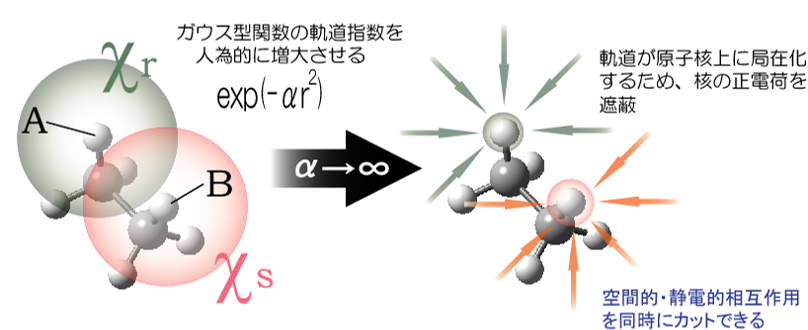
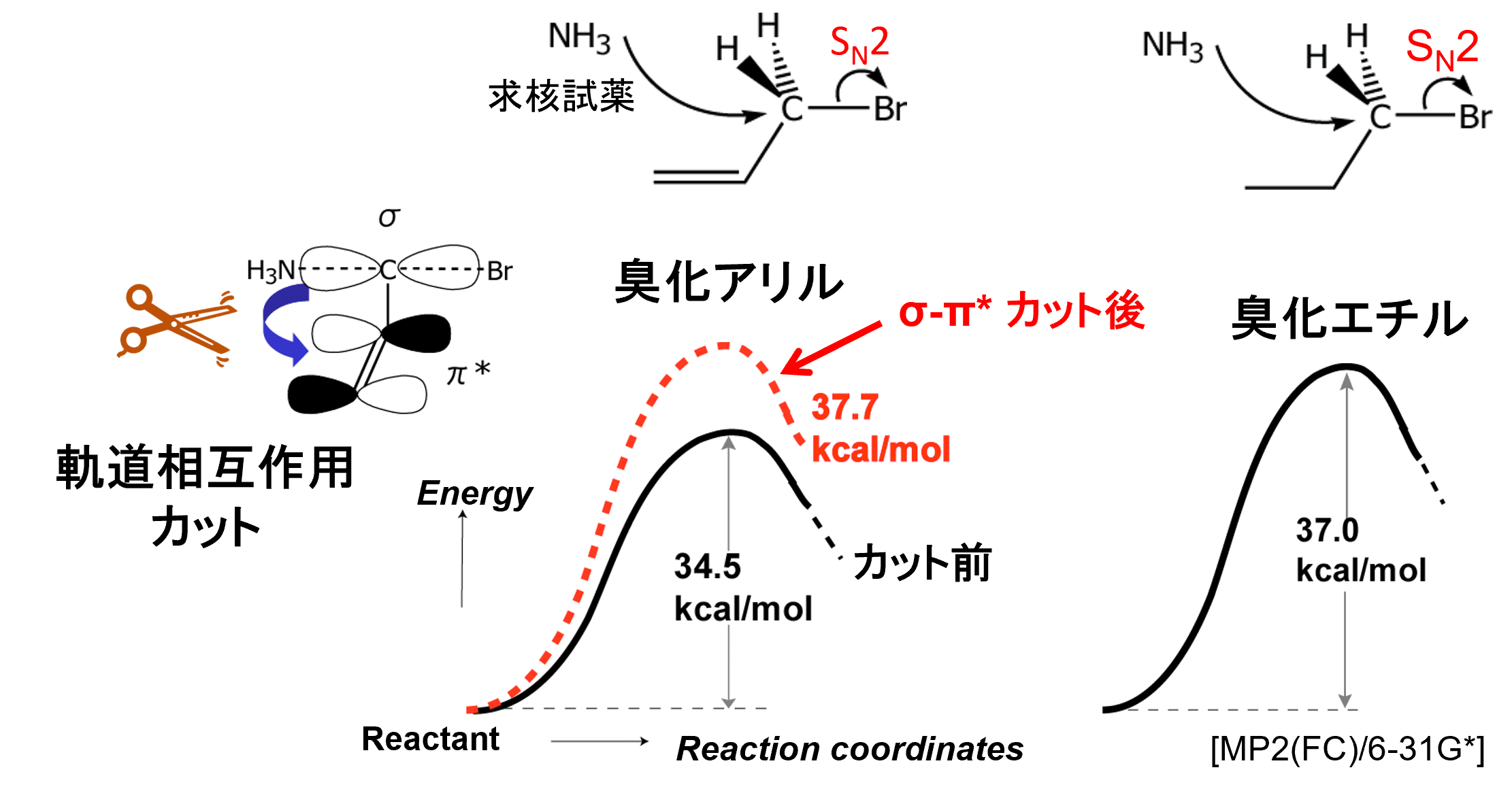
Through Space/Bond(TS/TB)解析法とは われわれは、相互作用の経路と機能発現の関係を、数値的な根拠をもって解釈できる定量的な解析手法を開発しています。TS/TB法[4,5]は、人為的軌道収縮により、特定の相互作用を電子-電子間、電子-核間、核-核間相互作用をバランスよくカットし、分子内・分子間の軌道間相互作用エネルギーを、電子相関効果を含めて定量的に評価可能な解析法です。本方法により、分子の安定性、機能と分子構造の関係、電荷移動、相互作用伝搬経路などの理論的解明が可能です。
解析例 (TS/TB) 臭化アリルのSN2反応は臭化エチルよりも100倍以上速く進むことが知られています。下図の黒線のように臭化アリル(34.5kcal/mol)は臭化エチル(37.0kcal/mol)よりも活性化エネルギー(Ea)が小さいことが量子化学計算から示され、実験を支持しています。しかし『なぜ臭化アリルのEaが低いのか』に対する答えは通常得られません。そこでTS/TB解析法で臭化アリルのπ-σ*相互作用をカットして当該相互作用の無い仮想状態を作り出したところ、赤線のように臭化アリルのEaが37.7kcal/molとなり(+3.2kcal/mol増加)、臭化エチルに近づきました。これにより、臭化アリルのπ-σ*相互作用がSN2反応の遷移状態を3.2kcal/molほど安定化させていることが低いEaの原因という定量的な説明ができます[5]。
『なぜ』を定量的に説明できれば、化学反応を含む諸現象や物質機能の発現機構を突き止めることが可能となり、従来アプローチを超えた革新的な創薬・材料開発に直接的な貢献が可能となります。